
Extractables & Leachables
What are Extractables & Leachables (E&L)?
Extractables are chemicals that extract from components of a manufacturing or packaging system into a solvent under forced conditions.
Leachables are compounds that can migrate via contact with manufacturing systems, container-closure systems, and drug delivery device components into the drug product.
E&L Study Design
The first step in an E&L study design is to collect information regarding the composition of the product, the delivery system, the packaging materials, and the intended use conditions. This is followed by a safety risk assessment. Different extraction procedures and techniques are then performed to evaluate toxicity. These toxicological assessments define the safety concern threshold levels for the extracted compounds.
The identification of extractables involves extensive data interpretation, library searches, data comparisons, confirmation by several analytical techniques, and analytical expertise. The quantitation of any potential leachables is undertaken based on the substances of concern that have been identified through extractables study. Toxicity quantification and assessment of any detected leachables that are above the analytical evaluation threshold will determine the overall product safety.
Limits for E&L
- Safety concern threshold – total daily intake threshold below which a leachable presents negligible safety concerns from carcinogenic and noncarcinogenic toxic effects
- Analytical evaluation threshold – concentration above which E&L need to be identified and or quantified
About Impurities
What is an Impurity?
"Any component of the new drug substance that is not the chemical entity defined as the new drug substance" as defined by ICH guidelines.
Where do Impurities come from?
- API synthesis
- API purification
- Packaging & Storage (Extractables and Leachables)
Classification of Impurities
![]()
Organic Impurities [1]
Result during the manufacturing process and/or storage of the new drug substance. They can be identified or unidentified, volatile or non-volatile.
- Starting ingredient reagents
- Reagents and catalysts
- Process by product
- Process intermediates
- Degradation products
- i.e., PAHs, Nitrosamines, Azo
- Dyes, Vulcanizing Agents,
- Phthalates, antioxidants,
- Elastomers, Stabilizers

Inorganic Impurities [1]
Result from themanufacturing process. They are normally known and identified.
- Reagents, ligands, and catalysts
- Heavy metals (elemental impurities i.e., Hg, Pb, Cd, As) [2] or other residual metals
- Inorganic salts
- Other materials (e.g., filter aids, charcoal)
![]()
Residual Solvent Impurities [3]
Inorganic or organic liquids used for the preparation of solutions or suspensions in the synthesis of a new drug substance. They are of known toxicity.
- Any chemical used in the synthesis process (i.e. benzene, methanol)
1 - efer to ICH Q3A Impurities in New Drug Substances / Q3B Impurities in New Drug Products
2 - refer to ICH Q3D Guideline for Elemental Impurities
3 - refer to ICH Q3C Maintenance of the Guideline for Residual Solvents
Other Impurities Include
- extraneous contaminants
- polymorphic forms
- enantiomeric impurities
About Impurities & Control
Why Control Impurities?

E&L Control
A controlled extraction study is a laboratory investigation into the qualitative and quantitative nature of extractable profiles from critical components of a container/closure system.
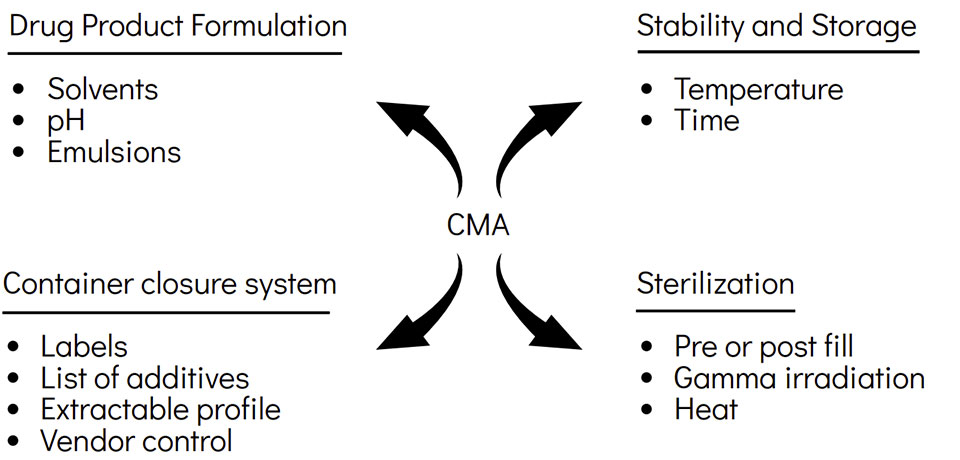
A critical material attribute (CMA) is a property or characteristic of an input material that should be within an appropriate range to ensure the desired quality of that drug substance, excipient, or in-process material. Below are the CMA's for E&L:
Dalton's Extractables and Leachables Services
As per U.S. FDA 21 CFR 211.94(a) and the European Commission Directive (2001/83/EC), regulatory standards necessitate pharmaceutical industries to develop sensitive and accurate analytical methods to detect, identify, and quantitate extractables and leacheables in a drug product.
With Dalton's new E&L consultation services and our customized testing capabilities, we can offer you a solution to your extractable and leachable needs. Our analytical team will be happy to help you overcome any concerns and challenges. Recently Dalton has overcome the complexity of E&L testing on drug products that contain phosphate ions.
Our solution: stable isotope labelled standards (SILs) to quantitate leachables in the sample matrix.
Dalton can also help you determine extractables and leachables on your next project:

Determination of the Analytical Evaluation Threshold (AET) based on Safety Concern Threshold (SCT) and dose per container.
Routine analysis of extractables and leachables in drug product by chromatographic and spectroscopic techniques.
Development of tailored study designs for extractables and leachables, including profiling
(inorganic and organic) and simulated leachable studies.
Development of analytical methods to evaluate Chemical and structural changes (e.g. by DSC) in polymeric container closure systems by spectroscopic or thermal analysis
Validation of analytical methods developed for determination of extractables and leachables on container closure systems.
Learn more about Dalton's extractables and leachables testing services.
Learn more about Dalton's pharmaceutical development manufacturing services.
View this page in PDF format.
Extractables & Leachables Studies and Testing
The characterization of extractables provides medical-grade qualification and chemical characterization information which is critical to avoid drawbacks during development. These drawbacks may include the introduction of non-intentionally added substances (NIAS) and contaminants, or even compounds migrating from printing and adhesives into the drug products.
No single analytical technique in the study of extractables and leacheables can be used for all categories of compounds. As a result, testing for extractables and leachables is achieved by complementary use of a variety of analytical techniques:
Extractables & Leachables Regulations
Guidance Documents & Pharmacopeia
Some international authorities require very detailed information on specific impurities (e.g., Germany and France require viral clearance in biological products). Refer to the guidance documents and pharmacopeias around impurities and reporting levels.
US FDA
- Title 21 of the eCFR Part 211.65, states that “Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements ” requiring the assessment of extractables and leachables.
- Guidance for industry: container closure systems for packaging human drugs and biologics (1999)
EMA
HC
US Pharmacopeia
- Plastic packaging systems for pharmaceutical use
- Assessment of E&L associated with pharmaceutical packaging/delivery systems
- Assessment of drug product leachable associated with pharmaceutical packaging/delivery systems
- Orally inhaled nasal drug products
ISO Guidelines for Medical Devices
International Standard
Currently, there are no internationally harmonized guidelines on the assessment and control of E&L. This current gap generates uncertainty for industry and regulators, creating potential delays in the approval of regulatory applications.
To solve this, the ICH is expected to release a guideline in November 2024 on the control of E&L, Q3E EWG Impurity: Assessment and Control of Extractables and Leachables for Pharmaceuticals and Biologics.
The FDA, EMEA, and other regulatory authorities are taking an increased interest in the interactions of various drug delivery devices, pharmaceutical product containers, and medical devices with a drug product and/or patient.
CTD Impurity Reporting
Impurity Reporting Requirements
PHASE I PHARMACEUTICAL
Reporting Requirements
- The structure (or other identifier, if not structurally characterized), as well as the origin, should be provided in the drug substance impurity table
PHASE II/III PHARMACEUTICAL
Reporting Requirements
- The impurity name (or identifier), structure (if characterized), and origin should be provided in the table for all specified impurities
- Impurity levels for previously manufactured nonclinical and clinical batches may also be summarized
- Demonstrate removal of any reagents used in manufacture
PHARMACEUTICAL LAUNCH
Reporting Requirements
- Will include all identified impurities except for the ones negotiated to be dropped
- Information is provided within section 3.2.S.3.2 (Impurities)
PHASE I BIOLOGICS
Reporting Requirements
- Must clearly demonstrate the purity of your drug product within an established range
- Must clearly demonstrate all established controls to monitor manufacturing
PHASE II/III BIOLOGICS
Reporting Requirements
- Clearly establish a validation report to demonstrate viral clearance if applicable
- Raw material (animal sourced) certifications to claim your drug product is free from TSE material (Transmittable Spongiform Encephalopathy)
- Removal of any reagents from the manufacturing process
BIOLOGIC LAUNCH
Reporting Requirements
- Will include all identified impurities except for the ones negotiated to be dropped
- Information is provided within the developmental section of Module 3.2.S.3.2 (Impurities) and Module 3.2.A.2; Adventitious Agents Safety Evaluation (TSE certificates – Viral Clearance Validation)
CTD Amendments and Notifications
Required where a new impurity has been identified.
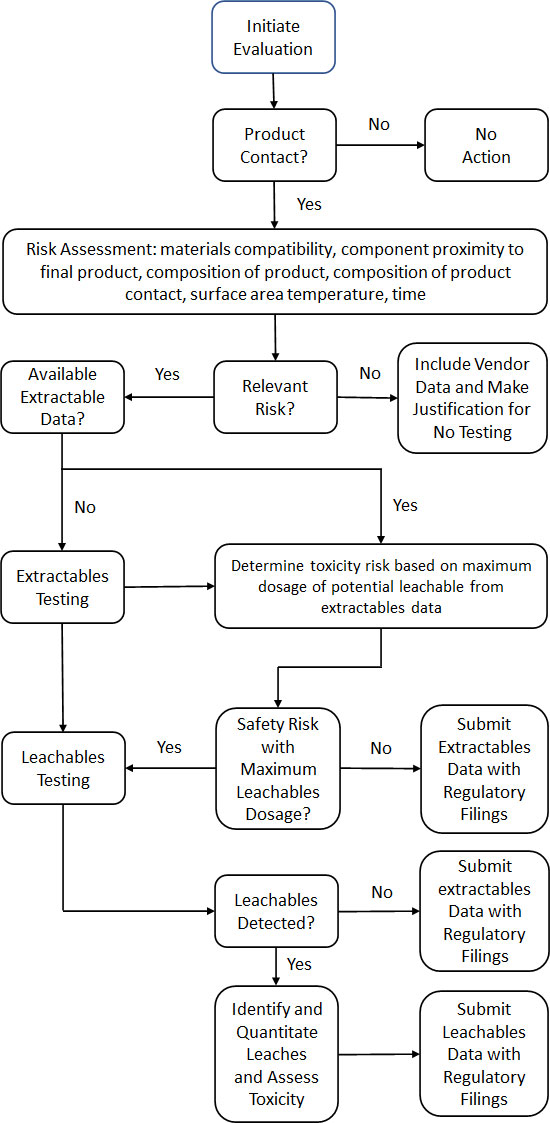
Extractables & Leachables Assessment Process
References
- Hotha, K. (2017). Extractables and leachables regulatory perspectives [7]
- International Conference on Harmonisation, Guideline for Impurities in New Drug Substances Q3A(R2)
- Li, K., Rogers, G., Nashed-Samuel, Y., Lee, H., Mire-Sluis, A., Cherney, B., ... & Markovic, I. (2015).
Creating a holistic extractables and leachables (E&L) program for biotechnology products.
PDA Journal of Pharmaceutical Science and Technology, 69(5), 590-619. - Yu LX, Amidon G, Khan MA, et al. Understanding pharmaceutical quality by design.
AAPS J. 2014;16(4):771-783. doi:10.1208/s12248-014-9598-3